Principles
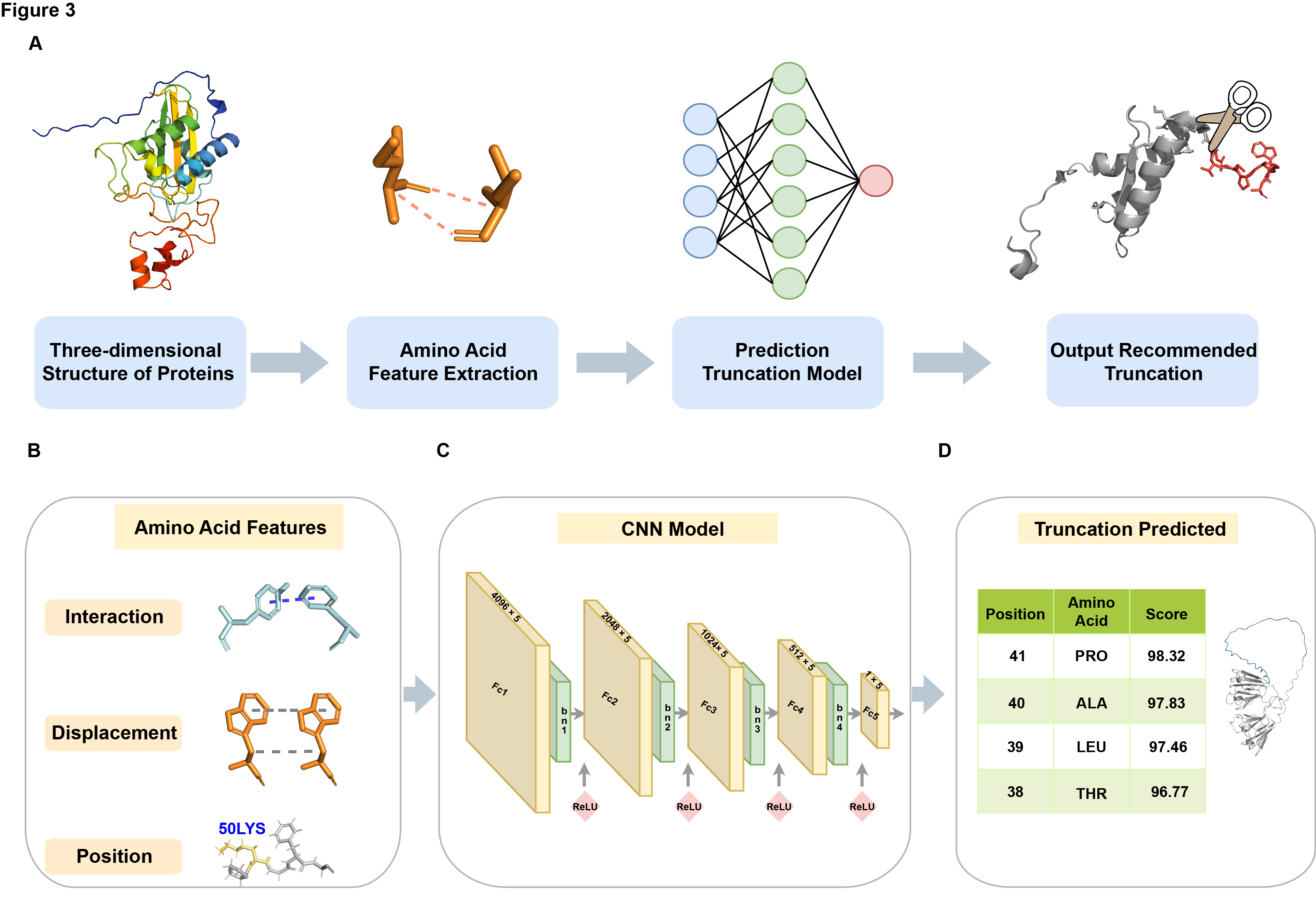
The overall workflow is illustrated in Figure 3. Initially, dynamic simulation results were aligned using PyMOL, with the first frame serving as the alignment center. Subsequently, our custom tool was employed to extract dynamic information for each amino acid in the protein, including relative displacement, interaction force duration, and positional information within the sequence. Finally, this dynamic information was input into our predictive model, which was trained using the dynamic data from eight proteins along with their corresponding optimal truncation points. The model then provided recommendations for the optimal truncation sites of the input protein, thereby assisting researchers in more accurately determining the necessary length for protein crystallization, significantly reducing workload and increasing efficiency.
Shanghai University of Traditional Chinese Medicine