DeepMAX
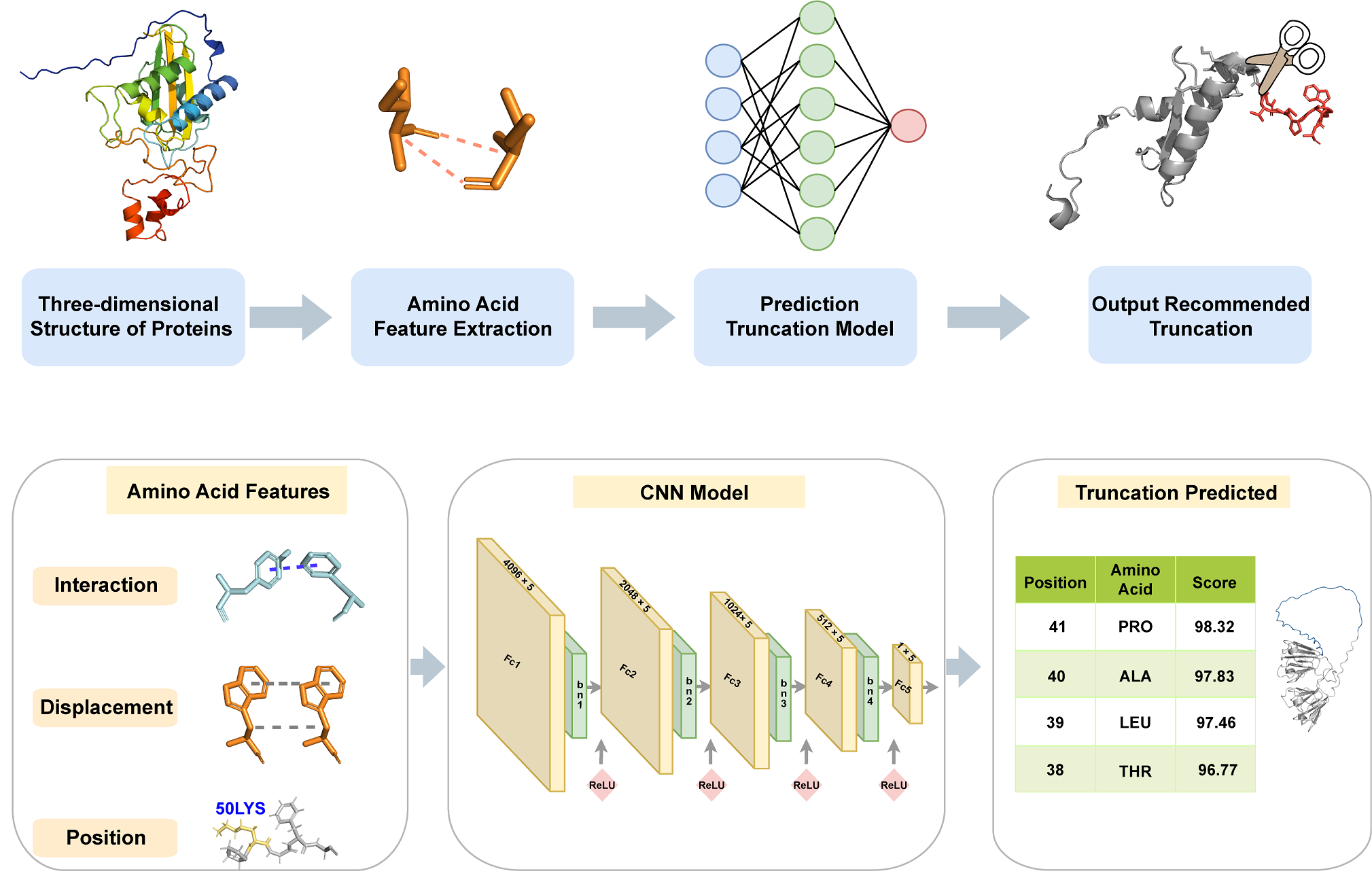
DeepMAX is a computational tool that integrates molecular dynamics simulations and artificial intelligence to optimize protein truncation for structural determination. High-resolution protein structures are essential for understanding biological functions and enabling structure-based drug design. Cryo-electron microscopy (Cryo-EM) and X-ray crystallography are among the most widely used techniques for protein structure determination. However, obtaining high-quality protein crystals remains a major challenge in X-ray crystallography. DeepMAX addresses this challenge by analyzing protein dynamic stability and providing detailed insights into the structural behavior of different regions, enabling informed truncation decisions. By incorporating root-mean-square fluctuation (RMSF) analysis and principal component analysis (PCA), DeepMAX identifies flexible regions that may hinder crystallization and suggests optimal truncation sites. This approach significantly improves experimental efficiency by reducing unnecessary steps, narrowing the search space, and minimizing resource and time consumption. By integrating molecular dynamics simulations, machine learning, and bioinformatics, DeepMAX provides a comprehensive framework for optimizing protein crystallization, ultimately enhancing the success rate of structural determination and facilitating structure-based drug discovery.
Citation
DeepMAX: Deep Learning for Protein Structure Optimization via MD, AlphaFold, and Crystallography
PMID : XXXXX